
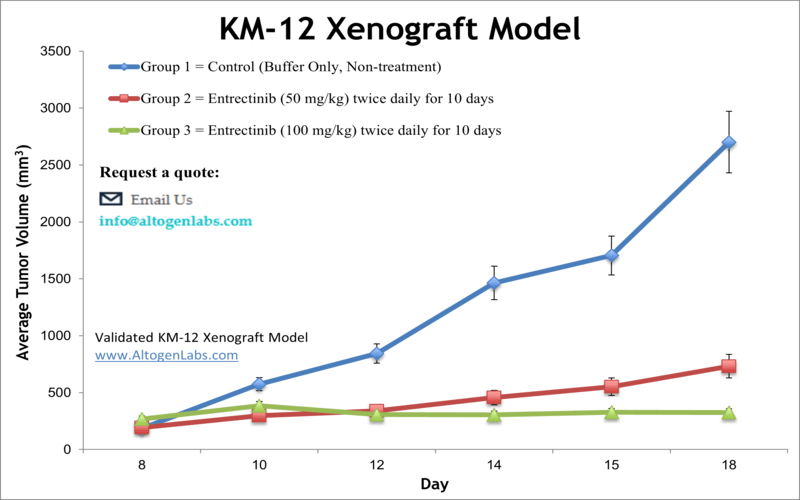
KM-12 xenograft model
Colorectal carcinoma remains a significant global health burden, representing the second leading cause of cancer-related mortality in males and the third in females. The KM-12 cell line, derived from a Dukes B2 stage colorectal carcinoma, has emerged as a valuable model for preclinical evaluation of therapeutic efficacy due to its well-characterized molecular phenotype and reproducible tumorigenicity in murine xenograft systems. KM-12 exhibits a relatively low metastatic potential and possesses a rare TPM3-NTRK1 gene fusion that results in constitutive activation of the TRKA kinase. This genetic alteration renders KM-12 cells highly sensitive to TRKA inhibitors, such as entrectinib and crizotinib, making the model a key asset in the study of NTRK-driven oncogenesis. The cell line has also demonstrated utility in evaluating drug responses in brain metastasis models through in vivo imaging methodologies. Combination therapy studies utilizing oxaliplatin and cetuximab have shown that cetuximab effectively reverses oxaliplatin resistance in KM-12 xenografts by attenuating downstream DNA repair pathways involving XRCC1, and by enhancing platinum-induced genotoxicity through inhibition of EGFR signaling. Furthermore, studies have confirmed the efficacy of NMS-P626, a selective TRKA inhibitor, in targeting KM-12 tumors bearing TPM3-NTRK1 fusion. The KM-12 cell line is routinely used to establish subcutaneous cell line-derived xenograft (CDX) models for assessing monotherapy responses as well as synergistic effects of combinatorial regimens, particularly in the context of targeted therapies and chemoresistance mechanisms. Its defined mutation profile and predictable tumor growth kinetics support its continued application in translational colorectal cancer research.
KM12 Xenograft Model: Download ![]()
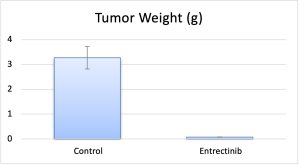
KM-12 Cell Line
The KM-12 cell line, derived from a human colorectal adenocarcinoma, has emerged as a pivotal model in cancer research due to its BRAF V600E mutation and KRAS wild-type genotype. This molecular profile renders KM-12 particularly relevant for investigating the efficacy and resistance mechanisms of targeted therapies involving the MAPK signaling cascade, such as BRAF and MEK inhibitors. Numerous studies have demonstrated that KM-12 cells exhibit constitutive activation of ERK signaling, which contributes to their oncogenic phenotype and intrinsic resistance to EGFR-targeted agents. Proteomic and transcriptomic analyses have revealed aberrations in cell migration, epithelial-mesenchymal transition, and PI3K/AKT signaling, highlighting their utility in studying metastatic behavior. KM-12 has also been extensively used in subcutaneous xenograft models and high-throughput drug screening to evaluate synthetic lethality in BRAF-mutant colorectal cancers. However, despite its widespread application, the model remains underexplored in 3D organoid systems and orthotopic implantation settings. Furthermore, the role of the tumor microenvironment, including stromal and immune components, in modulating therapeutic responses in KM-12 cells is insufficiently characterized, presenting a critical gap in the translational relevance of this model.
KM-12 Subcutaneous Xenografts in Colorectal Cancer Research
Subcutaneous xenograft transplantation represents a foundational technique in preclinical oncology, offering a standardized and accessible platform for assessing tumor growth and therapeutic response. The KM-12 cell line, a BRAF V600E-mutant colorectal adenocarcinoma model, has been widely utilized in such systems due to its reliable tumorigenicity and well-characterized molecular profile. When implanted into the flanks of immunodeficient mice, KM-12 cells consistently form tumors within a few weeks, allowing for reproducible evaluation of drug efficacy. These models have been instrumental in characterizing the effects of MAPK pathway inhibitors and elucidating mechanisms of adaptive resistance, particularly through upregulation of PI3K/AKT and RTK-mediated signaling pathways. Further research using KM-12 xenografts has demonstrated the therapeutic benefit of combinatorial approaches, such as co-inhibition of BRAF with PI3K or EGFR, which have produced superior antitumor effects compared to monotherapy. These studies have also enabled the identification of pharmacodynamic biomarkers like phosphorylated ERK and AKT to monitor treatment response. However, the subcutaneous model lacks the anatomical and microenvironmental fidelity of orthotopic systems, limiting its capacity to fully replicate tumor-stroma and immune interactions. Despite this, KM-12 subcutaneous xenografts remain powerful for mechanistic exploration and drug screening. Future advancements incorporating elements of the tumor microenvironment or humanized immune systems may enhance their translational relevance and broaden their applicability in modeling resistance and therapeutic outcomes.
Preclinical Efficacy of Merestinib in TPM3-NTRK1 Fusion-Positive Colorectal Cancer
In a study published by Konicek et al. in Oncotarget journal, the authors evaluated the preclinical efficacy of the multi-kinase inhibitor merestinib (LY2801653) in NTRK fusion-positive cancers, using the KM-12 colorectal carcinoma model as a central platform. Merestinib, an orally bioavailable type II inhibitor of MET, AXL, MKNK1/2, and NTRK1/2/3, showed potent inhibition of NTRK1 phosphorylation in KM-12 cells harboring a TPM3-NTRK1 fusion, with complete suppression of p-NTRK1 (Y490) at 62.5 nM. This translated to strong antiproliferative activity in both two-dimensional and three-dimensional cultures, with IC50 values of 10 nM and 45 nM respectively. In vivo, merestinib induced near-complete tumor regression in KM-12 xenografts and a patient-derived xenograft (EL1989) bearing the same fusion. The latter model showed a 39 percent reduction in tumor volume and a 63 percent decrease in Ki-67 staining, indicating substantial antiproliferative effects. Histological analysis revealed reduced tumor cell viability and notable mucin accumulation, potentially indicative of stromal or differentiation changes. To explore resistance mechanisms, the study utilized NIH-3T3 cells engineered to express TPM3-NTRK1 with G595R or G667C mutations. Merestinib retained efficacy against G667C-expressing tumors, producing sustained regression in vivo, while entrectinib showed only a transient response. Neither compound was effective against G595R-mutant tumors. X-ray crystallography confirmed that merestinib binds the NTRK1 kinase domain in a DFG-out conformation, avoiding steric interference from resistance-associated residues. These structural insights support merestinib’s activity in the setting of certain acquired mutations that confer resistance to type I inhibitors. While additional targets such as MKNK1/2 and their downstream effects on eIF4E phosphorylation may also contribute to antitumor activity, the study’s focus on KM-12 highlights merestinib as a promising therapeutic candidate for NTRK-driven malignancies, particularly those resistant to currently available NTRK inhibitors. Further clinical investigation is warranted.
Targeting the Mevalonate Pathway to Overcome KM12 Drug Resistance
Resistance to tyrosine kinase inhibitors in NTRK fusion-positive colorectal cancer is increasingly attributed to non-genomic mechanisms, particularly metabolic reprogramming. In KM12 colon cancer cells, which harbor a TPM3-NTRK1 rearrangement, resistance to multiple TRK inhibitors is consistently associated with overexpression of HMGCS2, an enzyme that regulates ketogenesis and feeds into the mevalonate pathway. This upregulation occurs regardless of the phosphorylation status of TRK or the activation state of downstream MAPK components, implicating a TRK-independent mechanism of resistance. Functional knockdown of HMGCS2 restores sensitivity to TRK inhibitors, induces apoptotic signaling, and suppresses proliferation in resistant KM12 sublines. These effects are reversed by supplementation with mevalonolactone, confirming the central role of the mevalonate pathway. Furthermore, pharmacologic agents such as simvastatin and silibinin effectively potentiate TRK inhibitor activity by suppressing HMGCS2 expression or function, with synergistic antitumor effects observed in vitro and in xenograft models. These results position HMGCS2 as a key metabolic driver of therapeutic resistance and underscore the broader relevance of lipid biosynthetic pathways in colon cancer progression. Unlike resistance mechanisms involving secondary kinase domain mutations or reactivation of canonical MAPK signaling, HMGCS2-mediated resistance operates independently of AKT and ERK, revealing an alternative survival axis through cholesterol synthesis and small GTPase activation. The delayed resistance observed with statin co-treatment suggests a practical approach for extending the efficacy of TRK-targeted therapies in NTRK-driven tumors. Given the clinical accessibility of statins and the mechanistic clarity provided by these findings, further exploration in immune-competent and orthotopic KM12 models is warranted. Targeting the metabolic landscape of cancer cells offers a compelling strategy to overcome drug tolerance and improve therapeutic durability in molecularly defined colorectal cancers.
Next-Generation TRK Therapy Shows Potency in KM12 Xenografts
Zurletrectinib is a TRK inhibitor that exhibits enhanced potency and intracranial activity against NTRK fusion-positive cancers, including colorectal cancer driven by the TPM3-NTRK1 fusion present in the KM12 cell line. In both biochemical and cellular models, zurletrectinib displayed low-nanomolar IC50 values against TRKA, TRKB, and TRKC kinases, and significantly inhibited proliferation in KM12 cells. Compared to first-generation inhibitors like larotrectinib, zurletrectinib was effective at doses up to 30-fold lower in in vivo xenograft models. Notably, this compound retained efficacy against several known TRK resistance mutations, including the solvent-front G595R and the xDFG G667A substitutions. However, its activity diminished against the G667C mutant, which creates steric hindrance due to cysteine bulk. In xenograft models derived from KM12 cells, zurletrectinib achieved tumor regression with minimal toxicity at low oral doses. Pharmacokinetic profiling in rats revealed that it had superior brain penetration relative to selitrectinib and repotrectinib, a feature further validated in intracranial glioma models bearing TRK resistance mutations. Mice implanted with resistant tumors and treated with zurletrectinib exhibited significantly prolonged survival compared to other treatment arms. These patterns confirm the drug’s enhanced bioavailability, robust target engagement, and improved therapeutic profile in both extracranial and intracranial settings. The methodology, incorporating kinase assays, in vivo modeling, and molecular docking, provides a rigorous preclinical framework.
TrkA Activity in KM-12 Colorectal Cancer
KM-12 is a human colorectal cancer cell line driven by a TPM3-NTRK1 gene fusion, which leads to constitutive activation of the TrkA receptor tyrosine kinase. This oncogenic alteration makes KM-12 an important model for evaluating targeted therapies and imaging strategies that focus on Trk fusion proteins. In vitro studies using the fluorine-18-labeled PET tracer demonstrated time-dependent uptake in KM-12 cells, peaking at over 200 percent radioactivity per milligram of protein within 60 minutes. Blocking experiments with entrectinib and non-radioactive TRACK confirmed high-affinity binding to TrkA, as both compounds produced similar low-nanomolar IC50 values. In vivo imaging in KM-12 xenograft-bearing mice showed selective tracer accumulation in tumor tissue, with tumor-to-muscle ratios increasing over time. However, absolute tumor uptake remained modest, which may reflect either limited expression or reduced accessibility of the intracellular kinase domain in vivo.
Xenograft animal models are essential in the preclinical evaluation of anti-cancer therapeutics by enabling in vivo assessment of drug efficacy against specific tumor types. These studies involve the engraftment of tumorigenic cell lines into immunocompromised mice or rats, either subcutaneously or orthotopically, followed by monitoring of tumor growth and response to treatment. All clinically approved oncology drugs have undergone evaluation using such models, which are highly intricate and require careful consideration of the animal strain, cell line characteristics, administration route, dosing schedule, and detailed analyses including tumor growth kinetics, histopathology, and molecular profiling of mRNA and protein expression. Altogen Labs supports this critical phase of drug development by offering over 90 standardized Cell Line Derived Xenograft (CDX) models and more than 30 Patient Derived Xenograft (PDX) models. Additional services include the generation of stable cell lines with long-term gene silencing or protein overexpression, as well as gene and protein expression analysis using RT-PCR and the ProteinSimple WES system.
KM12 Xenograft Model: Download ![]()
Basic study design
- KM-12 cells are collected during exponential growth. Viable cells are counted with trypan blue and need a minimum of 98% viability to proceed.
- The suspension concentration is adjusted and each mouse, athymic BALB/C or NOD/SCID and 10-12 weeks old, receives a single subcutaneous injection. The flank of the hind leg per mouse receives an injection of one million cells of the Matrigel + KM-12 cells.
- Injections are palpated to determine tumor establishment. The study begins at an average tumor size of 50-150 mm3, as measured by calipers (digital).
- Randomization of mice into treatment groups and subsequent compound administration is performed following the treatment schedule.
- Tumors are measured (daily) and mouse weights are recorded (up to 3 times a week).
- Necropsy and collections are performed as tumor size reaches the study limit. Tumors are weighed and documented.
- Collected tissues can be can be snap frozen in LN2 or prepared for histology.
Get Instant Quote for
KM-12 Xenograft Model
Animal handling and maintenance at the Altogen Labs facility is IACUC-regulated and GLP-compliant. Following acclimatization to the vivarium environment, mice are sorted according to body mass. The animals are examined daily for tumor appearance and clinical signs. We provide detailed experimental procedures, health reports and data (all-inclusive report is provided to the client that includes methods, results, discussion and raw data along with statistical analysis). Additional services available include collection of tissue, histology, isolation of total protein or RNA and analysis of gene expression.
Following options are available for the KM-12 xenograft model:
- KM-12 Tumor Growth Delay (TGD; latency)
- KM-12 Tumor Growth Inhibition (TGI)
- Dosing frequency and duration of dose administration
- Dosing route (intravenous, continuous infusion, intraperitoneal, intratumoral, oral gavage, intramuscular, subcutaneous)
- KM-12 tumor immunohistochemistry
- Blood chemistry analysis
- Toxicity and survival (optional: performing a broad health observation program)
- Gross necropsies and histopathology
- Positive control group employing dox or cyclophosphamide, at a dosage of 10-50 mg/kg administered by i.v. or intramuscular injection